Обмен фенилаланина и тирозина
Фенилаланин относится к незаменимым аминокислотам, поскольку ткани животных не обладают способностью синтезировать его бензольное кольцо.
В организме фенилаланин используется только в синтезе белков, весь неиспользованный запас аминокислоты превращается в тирозин. В этом непосредственно участвует фермент фенилаланин-4-монооксигеназа (фенилаланингидроксилаза), обеспечивающий окисление ароматического кольца. Кофермент тетрагидробиоптерин в реакции окисляется до дигидроформы. Восстановление кофермента осуществляет дигидробиоптерин-редуктаза со своим коферментом НАДФН.
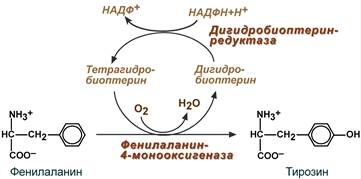
Реакция превращения фенилаланина в тирозин
Тирозин, помимо участия в синтезе белков, является предшественником гормона надпочечников адреналина, медиаторов норадреналина и дофамина, гормонов щитовидной железы тироксина и трийодтиронина и пигмента меланина.
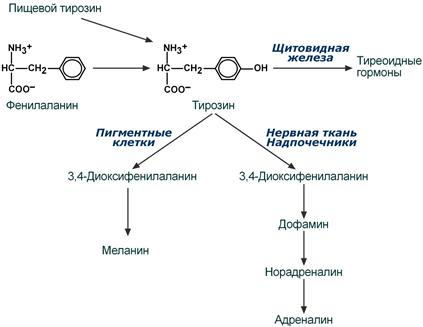
Пути превращения тирозина
Фенилкетонурия - самое яркое нарушение обмена аминокислот
При любых нарушениях превращения фенилаланина в тирозин развивается фенилкетонурия.
По Mc Kusick выделяется несколько типов фенилкетонурии: классическая (1 типа), вариантная (2 типа), 3 типа и материнская.
Фенилкетонурия 1 типа (классическая)
Фенилкетонурия 1 типа является наиболее распространенной аминоацидопатией. Частота ФКУ среди новорожденных по данным массового скрининга в различных странах составляет в среднем 1:10000, однако значительно варьирует в зависимости от популяции: от 1:4560 в Ирландии, до 1:100000 в Японии.
Этиология
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией, которая вызывает снижение активности фермента фенилаланин-4-монооксигеназы, обеспечивающей превращение фенилаланина в тирозин. Фермент имеется только в печени, почках, поджелудочной железе.
Патогенез
В патогенезе ФКУ имеют значение многие обстоятельства, в частности:
- значительное накопление в тканях и жидкостях больного организма фенилаланина и его производных (фенилпировиноградная, фенилмолочная (миндальная), фенилуксусная, гиппуровая кислоты, фенилэтиламин, фенилацетилглютамин) и вызванный ими ацидоз,
- прямое токсическое действие указанных веществ на центральную нервную систему, которое заключается в торможении фенилаланином активности ряда ферментов, в том числе пируваткиназы (окисление глюкозы), тирозиназы (синтез меланина), тирозин-гидроксилазы (синтез катехоламинов) и нарушение синтеза моноаминовых нейромедиаторов – тирамина, октопамина,
- нарушение синтеза серотонина, т.к. фенилаланин-4-монооксигеназа также вовлечена в гидроксилирование триптофана до 5-гидрокситриптофана, предшественника серотонина,
- конкурентное снижение фенилаланином транспорта в клетки ароматических аминокислот – триптофана и тирозина,
- нарушение синтеза простых и сложных белков в тканях, что вызывает тяжелые повреждения мозга и нарушение функции печени у большинства больных.
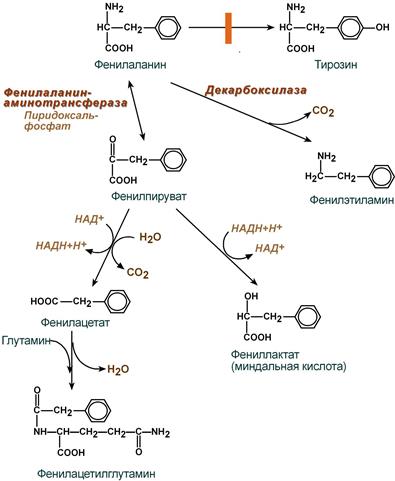
Превращение фенилаланина при фенилкетонурии
Клиническая картина
Ребенок с фенилкетонурией выглядит при рождении здоровым. Манифестация ФКУ происходит на первом году жизни, обычно в возрасте 2-6 мес. Первым симптомом заболевания может стать рвота. Другими ранними проявлениями болезни служат вялость ребенка, чрезмерная сонливость, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, плаксивость, также отмечаются срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), судороги.
Характерным признаком является повышенная потливость, от мочи и пота исходит необычный запах фенилуксусной кислоты, который характеризуют как заплесневелый, мышиный или волчий.
Дети отстают в физическом и нервно-психическом развитии.
NB! В связи с высокой частотой фенилкетонурии, тяжестью ее проявлений и возможностью профилактического лечения в России принято неонатальное обследование детей в роддомах в возрасте 4-5 дней.
У больных детей задержка психического и речевого развития может происходить постепенно и стать очевидной лишь через несколько месяцев. Нелеченный ребенок теряет около 50 баллов IQ к концу первого года жизни. Только 4-5% остаются на стадии дебильности, остальные имеют коэффициент интеллекта <60 и являются имбецилами или идиотами.
В старшем возрасте нелеченные дети становятся гиперактивными, осуществляют бесцельные движения, ритмические покачивания, у них определяется атетоз (мучительные непроизвольные движения кистей рук, языка, лица и др.). Дети имеют глубокую задержку моторного развития – 30% не ходит, 60% не говорит.
Основы лечения
Вовремя начатое лечение (диетотерапия) обеспечивает хороший клинический эффект, нормальную продолжительность жизни.
Единственным методом лечения является диетотерапия – исключение из питания больного высокобелковых продуктов питания с высоким количеством фенилаланина (мясо, рыба, яйцо, молоко, крупы). Вместо натурального белка используют специальные гидролизаты белка, частично или полностью лишенные фенилаланина.
NB! Однако больной должен получать с пищей определенные количества фенилаланина, покрывающие минимальную суточную потребность, что составляет 50-60 мг/кг для детей первого года жизни и 15-40 мг/кг для детей более старшего возраста. Это количество фенилаланина поступает за счет овощей, фруктов и других продуктов. Нельзя полностью исключать из рациона новорожденного материнское молоко, так как оно обеспечивает иммунитет ребенка в первые 6 месяцев жизни.
Строгое ограничение белков животного происхождения требуется на протяжении первых 2-3 лет жизни или как минимум до 6 лет, 5-10 лет, или до периода полового созревания (по разным данным). Во время беременности больные женщины должны возвращаться к диете, чтобы не допустить развития у ребенка умственной отсталости.
Больные нуждаются в дополнительном введении витаминов, особенно группы В, минеральных веществ и микроэлементов.
Фенилкетонурия 2 типа
Этиология
Аутосомно-рецессивный дефект дигидробиоптеринредуктазы.
В результате недостаточности фермента нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора гидроксилаз фенилаланина и триптофана. Вследствие этого нарушается превращение фенилаланина в тирозин, триптофана в 5-гидрокситриптофан.
Патогенез
Отмечается снижение уровня фолатов в сыворотке крови, эритроцитах и цереброспинальной жидкости. Это объясняется тесной взаимосвязью обмена фолатов и биоптерина, в частности участием дигидробиоптеринредуктазы в метаболизме тетрагидрофолиевой кислоты.
Клиническая картина
В клинической картине преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная дистония и гипотония, хореиформные движения (непроизвольные трясущиеся движения головы, лица или конечностей), спастический тетрапарез.
Течение болезни прогрессирующее и нередко приводит к смерти в 2-З-летнем возрасте. Появление клинической симптоматики, как правило, развивается в начале второго полугодия жизни, не смотря на диетотерапию.
Основы лечения
В отличие от классической формы этот вариант не поддается лечению ранним ограничением содержания фенилаланина в пище
Лечение тетрагидробиоптерином неэффективно, так как он не проникает через гематоэнцефалический барьер. Заместительная терапия L-ДОФА и 5-гидроокситриптофаном частично обходит блок в синтезе дофамина и серотонина.
Фенилкетонурия 3 типа
Этиология
Заболевание наследуется аутосомно-рецессивно и связано с недостаточностью 6‑пирувоил-тетрагидроптеринсинтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптерин-трифосфата.
Патогенез
Ключевую роль в патогенезе играет нарушение синтеза тетрагидробиоптерина. Развивающиеся при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ II.
Клиническая картина
Неврологические нарушения, в частности мышечная гипотония и задержка двигательного развития, появляются раньше, чем при классической ФКУ. Даже при адекватном снижении уровня фенилаланина в крови с помощью диеты, нарастание клинической симптоматики не прекращается. В отличие от больных ФКУ-II у этих больных судорог не отмечается (причина не ясна).
Основы лечения
Лечение тетрагидробиоптерином неэффективно, так как он не проникает через гематоэнцефалический барьер. Заместительная терапия L‑ДОФА и 5‑гидроокситриптофаном частично обходит блок в синтезе дофамина и серотонина. Диета неэффективна.
Другие варианты ФКУ
Также известны другие формы атипичной ФКУ, связанные с дефицитом тетрагидробиоптерина.
Недостаточность гуанозинтрифосфат-циклогидролазы описана по крайней мере у пяти больных. Этот фермент катализирует первую ступень синтеза тетрагидробиоптерина из ГТФ и при его дефиците в моче обнаруживается крайне низкая концентрация всех птеринов.
Материнская ФКУ
Этиология
Появление умственной отсталости среди потомства женщин с ФКУ, не соблюдающих диету в зрелом возрасте, получило наименование материнской ФКУ.
Патогенез
Патогенез патологии мало изучен, однако предполагается, что он сходен с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина в плазме крови матери. И в связи с накоплением этой аминокислоты в плаценте ее содержание в организме плода оказывается выше, чем у матери. Тем не менее, прямое токсическое действие фенилаланина точно не подтверждено.
Нарушения катаболизма тирозина
Тирозин, помимо участия в синтезе белков, является предшественником гормонов надпочечников адреналина, норадреналина, медиатора дофамина, гормонов щитовидной железы тироксина и трийодтиронина, пигментов. Нарушения катаболизма тирозина многочисленны и называются тирозинемии.
Тирозинемии.
Тирозинемия 1 типа
Этиология
Тирозинемия типа I (гепаторенальная тирозинемия) возникает при недостаточности фумарилацетоацетат-гидролазы. При этом накапливается фумарилацетоацетат и его метаболиты (сукцинилацетон), поражающие печень и почки. Частота заболевания 1:100-120 тыс новорожденных.
Клиническая картина
Существует две формы – острая и хроническая.
Острая форма составляет большинство случаев этой тирозинемии с началом в возрасте 2-7 мес и смертью 90% больных в возрасте 1-2 года из-за недостаточности печени.
К симптомам относится гипотрофия, рвота, «капустный запах» от тела и мочи, задержка развития, кровоточивость, диарея, мелена, гематурия, желтуха, анемия, периферические невропатии и параличи, кардиомиопатия, слабость мышц, дыхательные нарушения. Отмечают гипогликемию вследствие гиперплазии островковых клеток поджелудочной железы.
При хронической форме болезнь развивается позднее, медленнее прогрессирует. Продолжительность жизни около 10 лет.
Наблюдаются гипотрофия, узелковый цирроз печени и печеночная недостаточность, множественные дефекты почечной канальцевой реабсорбции с появлением синдрома Фанкони (щелочная рН мочи, глюкозурия, протеинурия), аминоацидурия, лейкопения, тромбоцитопения.
Из-за поражения печени и почек возникают проявления рахитоподобных заболеваний (остеопороз, остеомаляция). В результате печеночной недостаточности возникают симптомы, напоминающие острую порфирию. Непостоянными признаками являются умственная отсталость и неврологические изменения.
Основы лечения
Лекарственным средством является нитизинон, конкурентный ингибитор 4-гидроксифенилпируват-диоксигеназы. В результате прекращается образование гомогентизиновой кислоты и ее дальнейший распад. Также используется диета со снижением количества фенилаланина и тирозина, инъекции глутатиона.
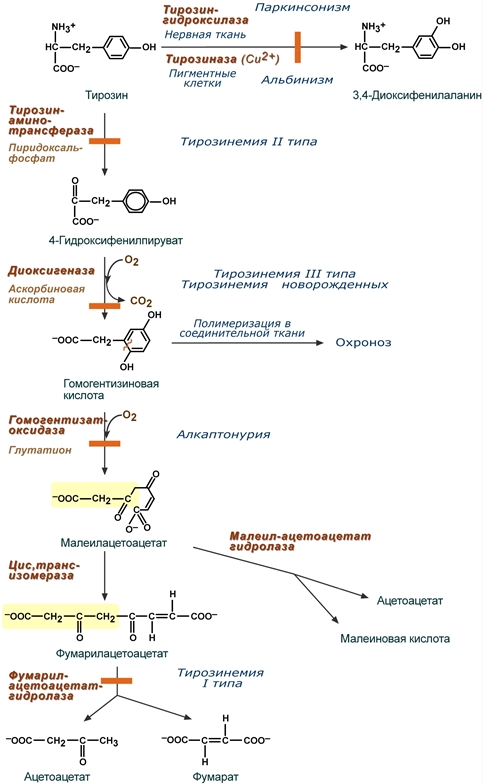
Дефектные ферменты и их реакции при тирозинемиях,
альбинизме и паркинсонизме
Тирозинемия 2 типа
Гораздо более редкое заболевание по сравнению с тирозинемией I типа.
Этиология
Тирозинемия типа II (глазокожная тирозинемия) возникает при недостаточности тирозин-аминотрансферазы.
Клиническая картина
Наблюдается задержка умственного и физического развития, микроцефалия, катаракты и кератоз роговицы (псевдогерпетический кератит), гиперкератоз кожи, членовредительство, нарушение тонкой координации движений.
Поражения почек и печени не наблюдается.
Основы лечения
Эффективна диета с низким содержанием тирозина, при этом поражения кожи и роговицы быстро исчезают.
Тирозинемия 3 типа
Этиология.
Тирозинемия III типа – результат генетического дефекта 4-гидроксифенилпируват-диоксигеназы. Зафиксировано лишь несколько случаев этой болезни.
Клиническая картина.
Характерные особенности включают умеренную умственную отсталость, судороги и периодическую потерю равновесия и координации (прерывистая атаксия).
Тирозинемия новорожденных
Этиология
Тирозинемия новорожденных – результат кратковременного снижения активности 4-гидроксифенилпируват-диоксигеназы. Чаще наблюдается у недоношенных детей.
Клиническая картина
Наблюдается сниженная активность и летаргия. Аномалия считается безвредной. Дефицит аскорбиновой кислоты усиливает клиническую картину.
Основы лечения
Диета со снижением количества белка, фенилаланина, тирозина и высокие дозы аскорбиновой кислоты (100 мг/день).
Алкаптонурия
Этиология
Генетическая аутосомно-рецессивная энзимопатия, частота варьирует 1:250 тыс.-1:1 млн. В основе заболевания лежит снижение активности печеночного фермента гомогентизат-оксидазы, в результате в организме накапливается гомогентизиновая кислота.
Клиническая картина
Так как гомогентизат на воздухе окисляется и полимеризуется в меланиноподобное соединение, то наиболее частым и постоянным симптомом является темная моча, на пеленке и нижнем белье остаются темно-коричневые пятна. Другим образом в детском возрасте болезнь не проявляется.
С возрастом гомогентизиновая кислота, накапливается в соединительно-тканных образованиях, склерах и коже, вызывает шиферно-глубокий оттенок ушного и носового хрящей (охроноз), окрашивает одежду, контактирующую с потеющими участками тела (подмышки).
Из-за связывания гомогентизата с коллагеном ухудшается состояние соединительной ткани, что делает хрупкими хрящевые образования. После 30 лет развивается дегенеративный артрит позвоночника и крупных суставов (бедренные, коленные), межпозвонковые пространства сужены, снижается минеральная плотность костей. Может наблюдаться поражение почек и сердца.
Основы лечения
Хотя эффективные способы неизвестны, по аналогии с другими аминокислотными нарушениями рекомендуется с раннего возраста ограничить потребление фенилаланина и тирозина, что должно препятствовать развитию охроноза и суставных нарушений. Назначают большие дозы аскорбиновой кислоты для снижения связывания гомогентизиновой кислоты в соединительной ткани. Предлагается использовать препарат нитизинон, конкурентный ингибитор 4-гидроксифенилпируват-диоксигеназы.
Альбинизм
Этиология
Заболевание обусловлено полным или частичным дефектом синтеза фермента тирозиназы (частота 1:20000), необходимой для синтеза диоксифенилаланина и далее меланинов в пигментных клетках.
Клиническая картина
При полном отсутствии фермента наблюдается тотальная депигментация кожи, волос, глаз, причем окраска одинакова для всех расовых групп и не меняется с возрастом. Кожа не загорает, совершенно отсутствуют невусы, какие-либо пигментные пятна, развиваются фотодерматиты. Сильно выражены нистагм, светобоязнь, дневная слепота (т.к. имеется депигментация сетчатки и ускоренный распад родопсина), красный зрачковый рефлекс.
При частичной недостаточности фермента отмечаются светло-желтые волосы, слабо пигментированные родинки, очень светлая кожа.
Основы лечения
Рекомендуется использовать различные средства защиты от ультрафиолетовых лучей.
Паркинсонизм
Этиология
Биохимической причиной паркинсонизма (частота после 60 лет 1:200) является низкая активность тирозин-гидроксилазы или ДОФА-декарбоксилазы в нервной ткани, при этом развивается дефицит нейромедиатора дофамина и накопление тирамина.
Клиническая картина
Наиболее распространенными симптомами являются ригидность мышц, скованность движений, тремор и самопроизвольные движения.
Основы лечения
Требуется систематическое введение лекарственных аналогов дофамина и применение ингибиторов моноаминоксидазы.
Синтез меланинов
Меланин (от греч. μελανος melanos – черный) – это пигмент, представляющий собой смесь различных полимерных соединений. Различают эумеланины, феомеланины, смешанные из двух предыдущих, и нейромеланины.
Образование пигмента происходит в меланоцитах, для этого в них существуют специальные органеллы – меланосомы. Наполненные меланином меланосомы продвигаются к апикальной части клетки и мигрируют в кератиноциты.
Начальным и ключевым ферментом синтеза меланинов является медь-содержащий фермент тирозиназа, имеющий тирозин-гидроксилазную активность и ДОФА-оксидазную активность. Ее активность регулируется влияниями эйкозаноидов и эндотелина-1, которые реагируют на гормональные влияния и физические факторы (например, ультрафиолет, температура).
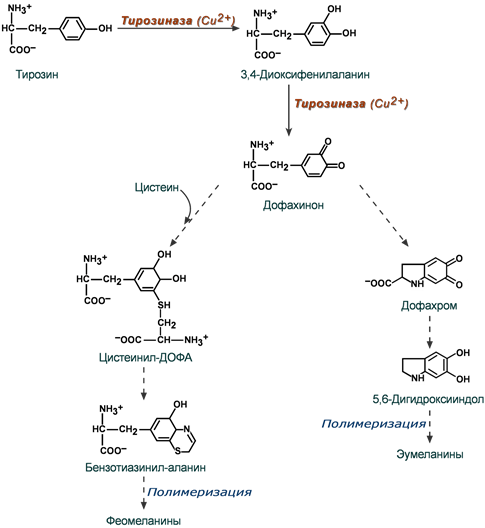
Схема основных этапов синтеза меланинов
Синтез меланинов начинается с превращения тирозина в диоксифенилаланин под влиянием тирозиназы. Далее этот же фермент образует дофахинон и здесь пути синтеза эумеланинов и феомеланинов расходятся. В синтезе феомеланинов и придании им теплых цветовых оттенков участвует цистеин.
NB! В животном и растительном мире, кроме цистеина, в синтез меланинов могут вовлекаться и другие аминокислоты – триптофан, гистидин, метионин, аргинин.Эумеланины обладают коричневым и черно-коричневым цветом, феомеланины – желтые или красновато-желтые. Эумеланины и феомеланины синтезируются меланоцитами кожи, сетчатки глаза и радужки, волосяных луковиц. За выработку меланинов в коже отвечает 4 или 5 генов (по разным данным), расположенных в разных хромосомах. Количество действующих генов определяет интенсивность синтеза меланина.
Нейромеланины образуются в дофаминергических нейронах черной субстанции головного мозга.
Функцией меланинов является защитная. Они поглощают ультрафиолетовые лучи, способны связывать катионы и анионы, хелатировать металлы (медь, марганец, хром, свинец, ртуть). Также меланины являются мощными антиоксидантами.
NB! Цвет волос изменяется в зависимости от количества, типа и распределения меланина в корковом слое. При увеличении количества пигмента он дополнительно может проникать в сердцевину волоса и усиливать темный оттенок. При низком количестве пигмента он распределяется диффузно и цвет волос будет светлым.
Цвет глаз определяется не только типом образуемого меланина, но также разным распределением его в слоях радужки и разным количеством. Если меланин присутствует только в четвертом и пятом слое, то у человека будет голубой или синий цвет глаз. При расположении пигмента в передних слоях радужки наблюдается карий или желто-коричневый цвет глаз, а при неравномерном распределении меланина в передних слоях – серый или зеленый
Обмен аспарагиновой и глутаминовой кислот
Аспарагиновая и глутаминовая кислоты являются отрицательно заряженными и заменимыми аминокислотами. Они легко образуются в клетках в реакциях трансаминирования, в которых оксалоацетат и α-кетоглутарат получают аминогруппы от других аминокислот.
В организме аспартат и глутамат используются всеми клетками для синтеза пуриновых и пиримидиновых азотистых снований. Амидные производные этих аминокислот являются транспортными формами аммиака из тканей в почки и печень. Кроме этого, глутаминовая кислота входит в состав глутатиона – вещества, выполняющего две различные функции – перенос аминокислот через мембрану и ключевое звено в антиоксидантной системе клетки. Также глутамат и его производное γ-аминомасляная кислота являются медиаторами в ЦНС.
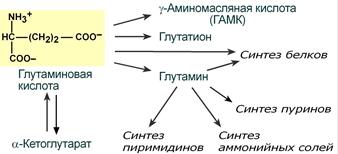
Пути использования глутамата
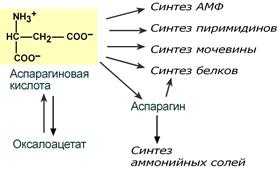
Пути использования аспартата
Обмен лейцина, валина, изолейцина и его нарушения
Валин, лейцин, изолейцин относятся к группе гидрофобных аминокислот, являются незаменимыми для человека и обладают разветвленным радикалом.
Аминокислоты активно участвуют в синтезе белков, особенно в мышечной ткани, играют роль в энергетике и метаболизме нервных клеток.
При распаде аминокислот они проходят ряд схожих этапов: трансаминирование с получением соответствующих α-кетокислот, их окислительное декарбоксилирование, еще одно окисление с образованием ненасыщенных кетокислот и уже более индивидуальные реакции превращения. Конечными продуктами распада являются для лейцина только ацетил-SКоА, для изолейцина и валина — ацетил-SКоА и сукцинил-SКоА.
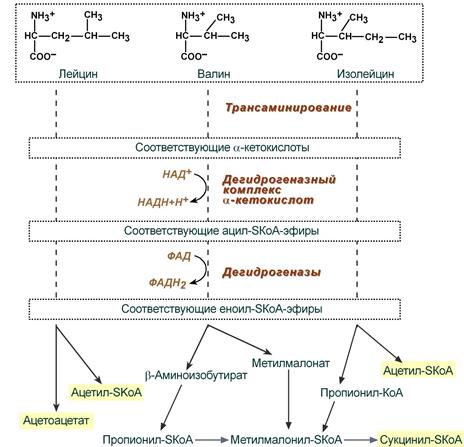
Этапы катаболизма лейцина, валина и изолейцина
(щелкните на схеме, чтобы увидеть химизм реакций)
Нарушения обмена этих аминокислот связаны с реакциями их катаболизма.
Лейциноз (болезнь мочи с запахом кленового сиропа)
Этиология
В основе заболевания лежит аутосомно-рецессивно наследуемый ферментативный блок окислительного декарбоксилирования кетокислот с разветвленной цепью, образующихся при распаде лейцина, изолейцина, валина. Эту реакцию осуществляет ферментативный комплекс дегидрогеназа α-кетокислот с разветвленной цепью. Частота примерно 1:180000 новорожденных.
Патогенез
До сих пор окончательно не выяснен. Но, так как известно, что лейцин активно поглощается нервной тканью, вероятно, нарушается его роль в энергетике нервных клеток и синтезе миелиновой оболочки. Обнаружено также понижение активности глутамат-декарбоксилазы и недостаточность образования ГАМК в мозге больных под влиянием повышенных количеств разветвлённых кетокислот.
Недоокисленные кетокислоты выделяются с мочой и придают ей специфический запах.
Клиническая картина
Клинически заболевание проявляется на первой неделе жизни рвотой, пронзительным криком и появлением характерного запаха мочи, напоминающего запах кленового сиропа, карамели, пережженного сахара или отвара овощей.
Одновременно появляется неврологическая симптоматика: отсутствие сухожильных рефлексов, мышечная гипотония, генерализованные и очаговые судороги, нарушение ритма дыхания. Отмечается замедленное психомоторное развитие, в дальнейшем – умственная отсталость. Возможно развитие коматозного состояния, ранний летальный исход.
Основы лечения
Лечение осуществляется только диетой с исключением соответствующих аминокислот.
Изовалератацидемия (болезнь с запахом потных ног)
Сходную с лейцинозом картину имеет и связанное с дефектом изовалерил-SKoA-дегидрогеназы изолированное нарушение обмена лейцина – изовалерат-ацидемия. Некоторым отличием от лейциноза является появление у больных запаха «потных ног», идущего от тела.
Обмен триптофана
Триптофан относится к группе гидрофобных ароматических аминокислот и для человека является незаменимой аминокислотой.
Поступающий в составе белков пищи триптофан в основном используется для биосинтеза белков, биогенного аминасеротонина и гормона мелатонина, и ниацина (витамин PP).
Метаболизм аминокислоты осуществляется в трех направлениях, которые сложны и на некоторых участках перекрещиваются друг с другом. Принципиально можно выделить следующие пути:
1. Кинурениновый (основной) – окисление и разрушение индольного кольца с образованием производных кинуреновой и антраниловой кислот. Большая часть триптофана распадается до ацетил-SКоА, но в одном из ответвлений этого пути одна из 60 молекул триптофана превращается в никотиновую кислоту (витамин B3, ниацин).
2. Серотониновый путь – окисление до 5-окситриптофана и далее превращение в серотонин и мелатонин.
3. Индольный путь – образование индольных производных, которые затем конъюгируются и выводятся с мочой.
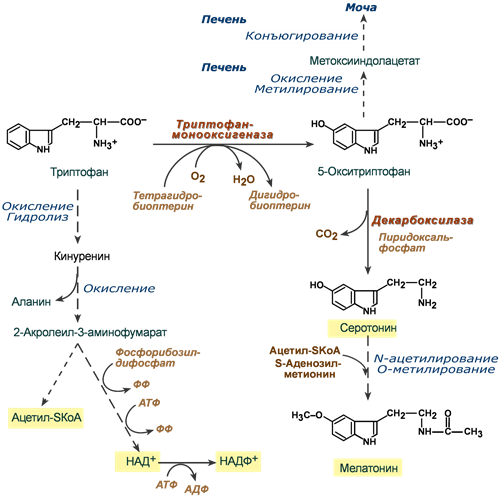
Реакции метаболизма триптофана
Синдром Хартнупа
Этиология
Синдром Хартнупа (имя больного, родители которого были двоюродными братом и сестрой) возникает в результате аутосомно-рецессивного дефекта транспортного белка для нейтральных аминокислот в эпителиальных клетках кишечника и почечных канальцев. Это приводит к снижению всасывания триптофана и ряда нейтральных аминокислот в слизистой кишечника и уменьшению их реабсорбции в канальцах почек.
Патогенез
В кишечнике триптофан подвергается воздействию микрофлоры с образованием производных (индолы, кинуренин и серотонин), которые всасываются и обнаруживаются в моче. В крови снижается концентрация триптофана, возрастает потеря нейтральных аминокислот с мочой, развивается гипераминоацидурия.
Так как триптофан необходим для синтеза эндогенного витамина РР, то клиническая картина характеризуется признаками пеллагры – недостаточности витамина B3 (PP, ниацина).
Клиническая картина
У пациентов поражается нервная система, кожа и слизистые оболочки, пищеварительная система и, соответственно, наблюдаются неврологические, психические и дерматологические проявления пеллагры, фоточувствительная кожная сыпь, эмоциональная лабильность, возможны энцефалопатия, преходящая мозжечковая атаксия, поражение печени и ЖКТ.
Одним из ярких проявлений синдрома является симптом голубых пеленок, возникающий из-за того, что избыток триптофана в кишечнике под действием микрофлоры превращается в индол, который всасывается в кровь и в печени обезвреживается до индикана. Далее индикан выводится с мочой и на воздухе окисляется в индиго и родственные ему соединения (индиготин) синего цвета.
Основы лечения
Симптомы болезни уменьшаются или даже исчезают при кормлении ребенка продуктами с высоким содержанием белка (4 г на 1 кг массы тела в день) и добавлением никотиновой кислоты (по 40-200 мг 4 раза в день).
Обмен серина и глицина
Серин и глицин являются полярными и заменимыми аминокислотами.
Роль реакции превращения серина в глицин состоит в образовании активной формы тетрагидрофолиевой кислоты – N5,N10-метилен-ТГФК.
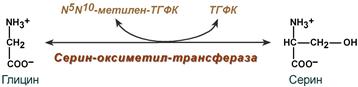
Реакция взаимопревращения глицина и серина
Одновременно данная реакция является первой на пути катаболизма серина. В других реакциях своего метаболизма серин
- при синтезе глюкозы подвергается неокислительному дезаминированию под действием фермента сериндегидратазы с образованием пирувата,
- на пути образования холина или бетаина декарбоксилируется,
- при образовании сфингозина – конденсируется с пальмитиновой кислотой.
Несмотря на простоту строения, глицин и серин являются весьма востребованными аминокислотами в клетках. Благодаря взаимопревращению перечень возможных путей метаболизма этих аминокислот еще больше расширяется.
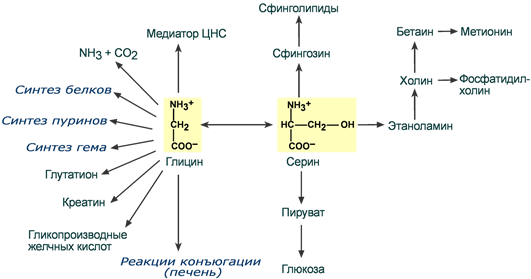
Пути использования серина и глицина
Роль метионина
Метионин является серусодержащей незаменимой аминокислотой, несущей ряд уникальных функций.
Первая функция заключается в инициации синтеза белка (трансляция). Взаимодействие метионина с первым кодоном мРНК является необходимым для создания первой пептидной связи будущего белка.
Вторая функция метионина основана на наличии в его структуре реакционноспособной метильной группы. Для того, чтобы ее активировать, к метионину присоединяется остаток аденозина и образуется S-аденозилметионин (SAM).
В результате реакций метаболизма эта метильная группа переносится на ряд субстратов. При этом образуются адреналин, мелатонин, креатин, карнитин, холин, фосфатидилхолин, гликозаминогликаны. Также S-аденозилметионин необходим для формирования 7-метилгуанозина («кэпа» на матричной РНК) – структуры, защищающей мРНК от преждевременного разрушения.
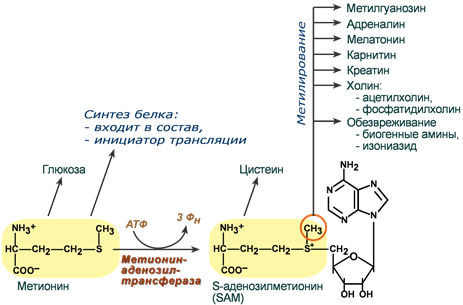
Пути использования метионина
Третьей функцией, благодаря все той же реакции метилирования, является способность метионина участвовать в обезвреживании биогенных аминов, детоксикации лекарств в печени (изониазид) и т.д.
Пути метаболизма цистеина
Цистеин является чрезвычайно важной аминокислотой в связи с тем, что это единственный источник органической серы для клеток организма.
В результате реакций метаболизма эта сера переходит в состав других серусодержащих веществ – фосфоаденозинфосфосерная кислота (ФАФС), коэнзим А, глутатион, сульфированные гетерополисахариды (хондроитинсульфат, кератансульфат, дерматансульфат) или выводится почками в виде сульфатов.
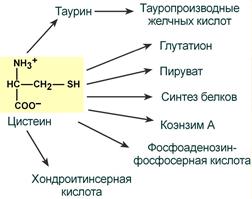
Пути использования цистеина
Одним из производных цистеина является таурин, обладающий разносторонними эффектами, механизм большинства которых малоизучен:
- является обязательным компонентом желчных кислот,
- играет роль внутриклеточного антиоксиданта, стабилизирует клеточные мембраны, оказывает кардиотропное действие.
- участвует в модификации митохондриальных тРНК, что влияет на митохондриальный синтез белка,
- стимулирует заживляющие процессы,
- выступает в качестве тормозного нейромедиатора.
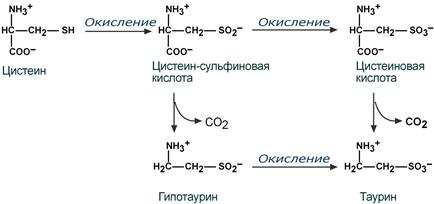
Реакции синтеза таурина
Связь обмена серина, глицина, метионина и цистеина
Образованный в реакции превращения серина до глицина N5,N10-метилен-тетрагидрофолат (активная форма витамина В9) при участии фермента метилен-ТГФК-редуктазы превращается в N5-метил-ТГФК. Его метильный остаток участвует в метионин-синтазной реакции реметилирования гомоцистеина в метионин.
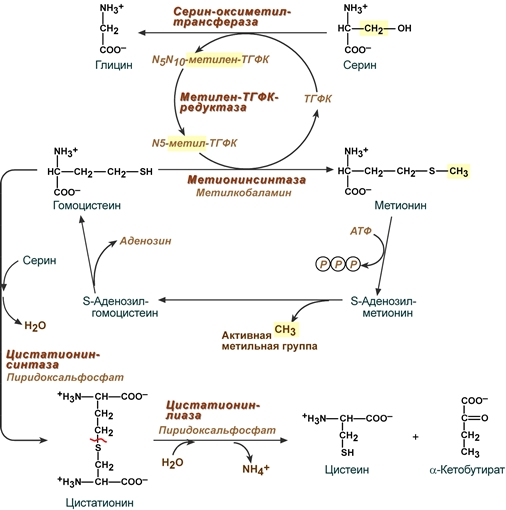
Взаимосвязь обмена серина, глицина, метионина и цистеина
Метионин впоследствии присоединяет аденозильный остаток и превращается в активную форму метионина – S-аденозилметионин, участвующий во многих реакциях метилирования, в частности, при синтезе креатина, карнитина, фосфатидилхолина, адреналина. В результате перемещения метильной группы и отщепления аденозина остается гомоцистеин, имеющий два пути метаболизма при нарушении которых развивается гомоцистеинемия:
Первый путь превращения гомоцистеина – реметилирование до метионина и вновь участие в реакциях метилирования и синтезе веществ.
Второй путь – взаимодействие с серином при участии цистатионин-синтазы, превращение в цистатионин с последующим распадом в цистеин и α-кетобутират.
NB! В печени, кроме метил-ТГФК, источником метильной группы может быть вещество бетаин (триметилглицин).

NB! Реакция превращения N5-метил-ТГФК в свободную ТГФК помогает удержанию ее в клетке, т.к. через клеточную мембрану она не проходит. При недостаточности витамина В12 N5-метил-ТГФК теряется из клеток и возникает внутриклеточный дефицит витамина В9.
Нарушение обмена метионина и цистеина
В настоящее время частым и очень актуальным нарушением является гомоцистеинемия – накопление гомоцистеина в крови из-за нарушения его метаболизма в метионин или цистеин.
Референсные величины гомоцистеина в сыворотке крови
| Дети | около 5 мкмоль/л |
| Подростки | 6-7 мкмоль/л |
| Взрослые | 5,0-15,0 мкмоль/л |
Повышение уровня разделяют на легкое (16-30), среднее (31-100) и тяжелое (>100 мкмоль/л).
NB! Понцентрации около 10 мкмоль/л были признаны ВОЗ пограничными при диагностике заболеваний, т.е. свыше этих показателей у людей, входящих в группу риска, можно утверждать о наличии патологии!
Этиология
Все причины гомоцистеинемии делят, как минимум, на две группы:
1. Самой распространенной причиной является недостаточность витаминов В12 (цианкобаламин), В6 (пиридоксин), В9 (фолиевая кислота), которые взаимодействуют с ферментами метионин-синтаза, цистатионин-синтаза, метилен-ТГФК-редуктаза, играющими центральную роль в метаболизме метионина и гомоцистеина.
2. Наследственный дефект указанных выше ферментов:
- гомозиготный (аутосомно-рецессивно) дефект цистатионин-синтазы (пиридоксин-зависимая гомоцистинурия), частота 1:100000, наблюдается повышение уровня общего гомоцистеина натощак до 40 раз.
Клинические проявления включают дислокацию хрусталика и другие глазные осложнения, проблемы с интеллектом примерно в 50% случаев, деформации скелета, ранний атеросклероз и сосудистые (атеротромботические) осложнения.
NB! Примерно у половины нелеченых гомозигот сосудистые осложнения наблюдаются до 30 летнего возраста. Гетерозиготная форма (примерно 1 на 150 человек в популяции) часто связана с нормальным базальным уровнем гомоцистеина, и пока не ясно, связана ли гетерозиготность с дополнительным риском сосудистых осложнений.
- чаще причиной наследственной гипергомоцистеинемии является гомозиготный дефицит метилен-тетрагидрофолат-редуктазы (пиридоксин-резистентная гомоцистинурия), при которой фермент имеет половинную активность от нормы (умеренная гомоцистеинемия).
NB! В белой популяции 10-13% людей являются гомозиготами по этой мутации, и при недостаточном потреблении витамина В9 у них повышается уровень общего гомоцистеина до 1,5 раз от нормы.
- нарушенная активность метионинсинтазы. Описано всего несколько случаев такого дефекта. Предполагается, что дефектным является фермент кобаламин-редуктаза, работа которого предшествует образованию дезоксиаденозил-кобаламина и метилкобаламина, т.к. одновременно наблюдается повышение концентрации метилмалоновой кислоты (симптом дефицита витамина B12).
Патогенез
Гомоцистеин, растворенный в плазме, провоцирует перекисное окисление липидов в липопротеинах крови и повреждение апобелков и тем самым вызывает нарушение связывания их с рецепторами и задержку их в крови. Особенно это ярко проявляется для ЛПНП, окисление которых приводит к их поглощению макрофагами в интиме и к активации атеросклероза. Параллельно гомоцистеин ускоряет агрегацию тромбоцитов и вызывает повреждение эндотелия сосудов, что интенсифицирует развитие тромбозов.
Сопутствующие заболевания
Гомоцистеинемия считается независимым фактором риска атеросклероза коронарных, периферических и мозговых сосудов (т.е. независимо от курения, уровня холестерина и артериальной гипертензии). Она обнаруживается в 30% случаев атеросклероза, тромбозов и ишемической болезни сердца. Также выявляется при болезни Альцгеймера, нарушениях беременности – невынашивание, мертворождения.
Основы лечения
При дефекте цистатионин-синтазы применяется лечение витамином В6 в дозе 250-500 мг/день. При дефекте метилен-тетрагидрофолат-редуктазы уровень гомоцистеина может быть снижен благодаря употреблению фолиевой кислоты по 5 мг/день. Витамин В12 также оказывает положительное влияние.
Одновременно назначается диета со сниженным содержанием метионина, что достигается специальным подбором продуктов, бедных этой аминокислотой.
NB! Повышенную склонность к гипергомоцистеинемиии имеют курящие. Выкуривание 20 сигарет в день приводит к увеличению концентрации гомоцистеина на 20 %.
Потребление больших количеств кофе является одним из самых мощных факторов, способствующих повышению уровня гомоцистеина в крови. У лиц, выпивающих более 6 чашек кофе в день, уровень гомоцистеина на 2-3 мкмоль/л выше (на 20-30 %), чем у не пьющих кофе. Предполагается, что негативное действие кофеина на уровень гомоцистеина связано с изменением функции почек.
Каждое повышение уровня гомоцистеина на 5 мкмоль/л сопровождается увеличением риска патологии мозговых артерий в 1,5 раза и периферических артерий в 6,8 раз. Увеличение концентрации гомоцистеина в крови более 22 мкмоль/л связано с 4-кратным повышением риска возникновения тромбоза глубоких вен.
У мужчин с уровнем ГЦ всего на 12% превышающим норму наблюдается тройное увеличение риска сердечного приступа.
Уровень гомоцистеина часто повышается при сидячем образе жизни. Умеренные физические нагрузки способствуют снижению его концентрации при гипергомоцистеинемии. Потребление небольших количеств алкоголя может снижать уровень гомоцистеина, а большие количества спиртного способствуют его росту в крови.
Цистиноз - это накопление цистина
Этиология
Аутосомно-рецессивная болезнь лизосомального накопления, обусловленная нарушением белка цистинозина, обеспечивающего транспорт цистина из лизосом. Возможно, также имеется энзиматический блок на пути его превращения, результатом которого является накопление цистиновых кристаллов в лизосомах. Частота – 1:100000 (в Англии и Франции – до 1:25000).
Патогенез
Происходит отложение цистиновых кристаллов в ретикулярных клетках костного мозга, в клетках печени, почек, селезёнки, слизистой оболочки прямой кишки, в лимфатических узлах и лейкоцитах, в клетках роговицы и конъюнктивы, в островковых клетках поджелудочной железы, аорте, атрофических яичниках и мозге.
Диагноз
Первыми симптомами являются полиурия, полидипсия, лихорадка неизвестного происхождения. При исследовании мочи выявляется щелочной рН, глюкозурия и протеинурия, что свидетельствует о синдроме нарушения функции почечных канальцев – синдроме Фанкони.
На втором году жизни ухудшается функция почек и развивается аминоацидурия с повышенным выведением цистина с мочой, обнаруживаются кристаллы цистина в моче (цистинурия).
Клиническая картина
Различают 3 клинические формы (проявляющиеся в раннем детстве, в юношеском возрасте и у взрослых).
Цистиноз
Цистиноз ранний нефропатический
Заболевание развивается на 1-м году жизни и при отсутствии терапии больные погибают от хронической почечной недостаточности до конца 1-го десятилетия жизни.
Отмечаются отставание массы тела, психического развития, часто развиваются анорексия, гипотрофия и потеря аппетита, отставание в росте, миопатия, общая мышечная слабость, расстройства памяти, атрофия мозга, часто возникает метаболический ацидоз, рвота, запоры, полиурия и полидипсия. Могут наблюдаться подъемы температуры тела, не связанные с инфекцией.
Изменяется функция почек, появляются признаки, указывающие на канальцевые повреждения. К концу первого года жизни нарушение почек вызывает развитие рахита и остеопороза. Увеличены в размерах печень и селезенка.
В роговице можно обнаружить кристаллические отложения и ретинопатию по типу «соли с перцем». Прогрессивно нарастает фотофобия. Кроме указанных отмечаются гипотиреоз, гипофункция яичников, эндокринная и экзокринная недостаточность поджелудочной железы.
Цистиноз нефропатический поздний
Отличается от раннего варианта нормальным ростом и началом в подростковом возрасте. Прогрессирует более медленно, однако прогноз также неблагоприятный.
Цистиноз доброкачественный взрослых
Начинается во взрослом возрасте. Характерно отсутствие аминоацидурии и нарушений функций почечных канальцев. Болезнь проявляется фотофобией, головными болями и слезотечением.
Основы лечения
Если больного не лечить, то наблюдается ранняя терминальная почечная недостаточность, тиреоидная недостаточность и мультиорганная дисфункция.
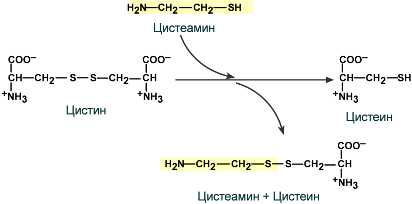
Введение цистеамина. Его молекула может реагировать с дисульфидной группой цистина, разрывая его структуру. При этом образуется молекула свободного цистеина и комбинация цистеин-цистеамин, которые выходят из лизосомы. Это снижает количество цистина в лизосоме и значительно улучшает течение заболевания. Постоянное применение цистеамина замедляет повреждение почек и других органов.
Трансплантация почки – имеет относительную эффективность, т.к. не устраняет причину, продолжается дегенерация внутренних органов вследствие накопления цистина.
Также применяют высокие дозы витамина D (100 000 ME в сутки), анаболические гормоны. Используется диетотерапия с ограничением белков, содержащих значимое количество серусодержащих аминокислот: метионина, цистеина и цистина.
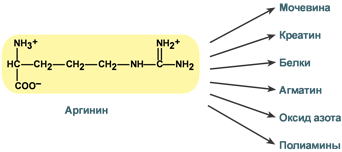
Роль аргинина
Аргинин является положительно заряженной и условно незаменимой аминокислотой. Понятие условно-незаменимая используется по той причине, что у детей и подростков, у пожилых людей синтез аргинина не покрывает потребности организма.
Аргинин в тканях входит в состав белков и, в частности, в большом количестве присутствует в гистонах, регулирующих активность ДНК. Метаболизм аргинина по аргиназному пути ведет к синтезу регуляторных полиаминов спермина и спермидина (см ниже). Превращение по NO-синтазному пути используется для образования оксида азота (NO), выполняющего функцию передатчика сигналов. Аргинин участвует в орнитиновом цикле синтеза мочевины и при синтезе креатина, выполяющего функцию запасного макроэрга.
NB! Продуктом α-декарбоксилирования аргинина является еще недостаточно изученный агматин, работающий как нейромедиатор. Он синтезируется в нейронах, хранится в синаптических везикулах, высвобождается путем деполяризации мембраны. Агматин связывается с α2-адренорецепторами и участками связывания имидазолина, блокирует рецепторы NMDA (N-метил-D-аспартат) и катионные каналы. Агматин снижает активность NO-синтазы (NOS) и, вероятно, индуцирует секрецию некоторых пептидных гормонов.
Пути метаболизма аргинина
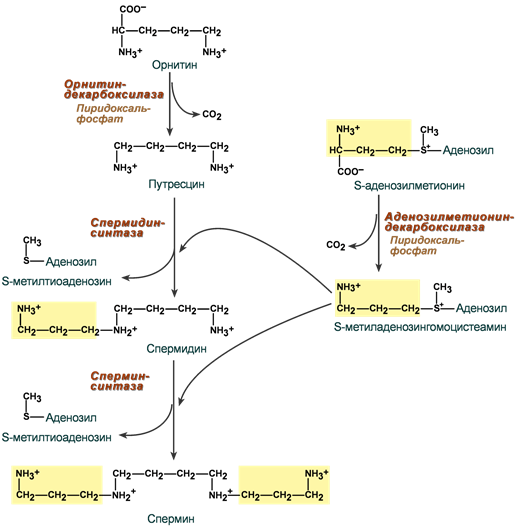
Полиамины
Действие аргиназы на молекулу аргинина приводит к образованию мочевины и орнитина. Орнитин в нескольких реакциях превращается в полиамины спермин и спермидин. Эти высокоактивные вещества содержатся в клетках всех типов и жизненно необходимы для их нормального роста и пролиферации.
Спермин и спермидин
- взаимодействуют с ДНК, РНК и нуклеопротеинами,
- служат регуляторами активности ферментов транскрипции, репликации и репарации,
- абсолютно незаменимы при синтезе одного из факторов инициации при трансляции,
- регулируют процесс полимеризации тубулина.
- участвуют в регуляции транспорта ионов Са2+ и ионов K+.
Синтез полиаминов спермина и спермидина
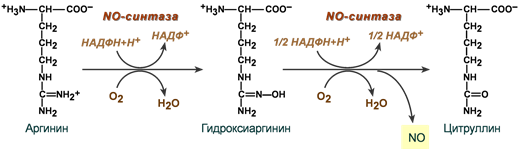
Оксид азота
Оксид азота образуется при ферментативном окислении L-аргинина. Процесс сложен и катализируется NO-синтазами (NOS), кофакторами в реакции выступают НАДФН, тетрагидробиоптерин, ФАД и ФМН.
Синтез оксида азота
(участие ФАД. ФМН, тетрагидробиоптерина не показано)
Оксид азота обладает широким спектром биологического действия, являясь незаряженной сигнальной молекулой, свободно диффундирующей между клетками:
- выступает как вторичный мессенджер и активирует цитозольную гуанилатциклазу,
- является нейромедиатором,
- играет роль в регуляции сосудистого тонуса и расслаблении гладкой мускулатуры сосудов,
- предотвращает агрегацию тромбоцитов и адгезию нейтрофилов к эндотелию,
- обладает цитотоксической и микробицидной активностью.
NB! В практическом здравоохранении широко используются нитратсодержащие противоангинальные препараты (нитроглицерин, нитросорбид, амилнитрит и др.), лечебный эффект которых обоснованно связывают с их способностью высвобождать в организме оксид азота.